


2026 VOL.126




AI가 찾아낸 희귀질환과 암의 ‘숨은 범인’
종결 코돈 변이 병원성 입증
진단검사의학과 윤지훈 교수
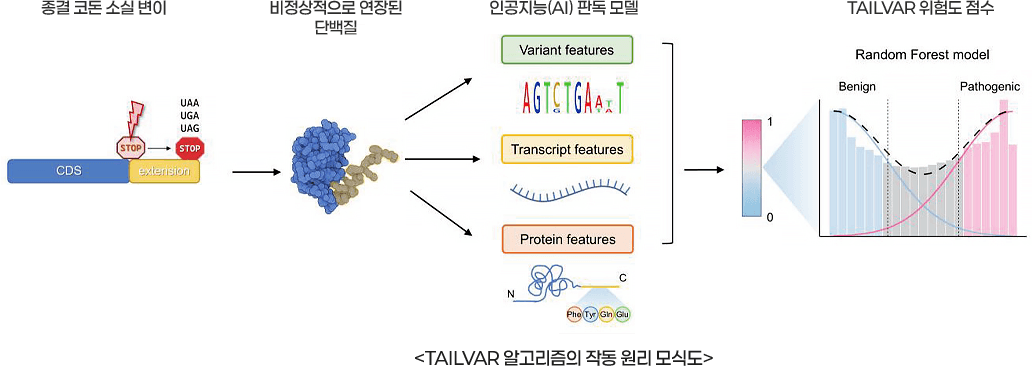
종결 코돈 변이는 단백질 합성을 조기에 멈추게 하거나 반대로 끝나야 할 지점이 사라지게 만드는 변이로, 유전질환에서 매우 중요하게 다뤄진다. 하지만, 그간 유전자 검사에서 판독이 까다로워 질병과의 연관성을 규명하지 못해 의미 불분명 변이로 남아 있었다. 이에 진단검사의학과 윤지훈 교수팀이 종결 코돈 변이의 병원성을 높은 정확도로 예측하는 AI 기반 유전자 변이 판독 모델 TAILVAR를 개발해 주목을 받고 있다.
정리 편집실 / 사진 송인호
이번 연구를 시작하게 된 배경은 무엇인가요?
강남세브란스병원은 의사과학자 양성을 위해 많은 노력을 기울이고 있습니다. 유전체 연구를 수행하는 연구자로서 이러한 환경에 자연스럽게 참여하게 되면서 한국보건산업진흥원의 ‘글로벌 의사과학자 양성사업’의 지원을 받아 이번 연구를 진행했습니다. 진단검사의학과에서는 진단이나 치료제 선택의 목적으로 처방된 유전자 검사들의 DNA 시퀀싱을 수행하고 판독합니다. 하지만, DNA 서열 정보가 어떤 임상적 의미가 있는지 아직 밝혀지지 않은 부분이 너무 많아 현재도 전 세계적으로 유전체 분야와 관련한 연구가 활발하게 진행되고 있습니다.
최근에는 유전자 염기서열 분석, 유전자 발현 패턴, 유전자 변이 탐지 등 다양한 유전체 연구에 AI 기술이 활용됨으로써 판독하기 어려운 유전자 변이를 새롭게 해석할 수 있는 가능성이 열렸습니다. 그간 아미노산이 치환되는 변이들에 대한 연구가 많이 진행됐지만, 종결 코돈 변이는 여전히 해석이 안 된 채 남아 있어 관심을 갖게 되었습니다.
이번 연구는 어떻게 진행되었나요?
종결 코돈(Stop codon)은 세 가지 염기의 조합(TGA, TAG, TAA)으로, 우리 몸속 세포의 단백질 합성을 멈추게 하는 일종의 ‘정지 신호’입니다. 정상적인 경우에는 이 신호에 맞춰 단백질이 일정한 길이로 만들어지지만, 유전자 서열에 변이가 생겨 이 신호가 사라지면 단백질 말단(C-terminal)이 비정상적으로 길어져 꼬리가 달린 단백질이 만들어집니다. 이렇게 변형된 단백질은 길어진 서열로 인해 세포 내에서 엉겨 붙어 독성을 유발하거나, 세포 보호를 위한 비정상 단백질 분해 시스템에 의해 제거됨으로써 본래의 기능을 잃게 됩니다. 그로 인해 결과적으로 도파민 반응 이상운동증, 뮤코다당증과 같은 희귀질환은 물론 유방암과 대장암 등 다양한 유전성 암을 유발하는 원인이 됩니다.
문제는 이러한 변이 중에는 실제 병을 일으키는 병원성 변이와 인체에 영향을 미치지 않는 양성 변이가 섞여 있는데, 이를 정확히 판독하기가 매우 어렵다는 것입니다. 이처럼 길게 늘어나는 단백질을 판독하기 위한 툴을 만들어보자는 목적으로 단백질 말단이 연장되는 종결 코돈 변이의 생화학적 특성에 주목했습니다.
연구 진행과 결과 도출 과정이 궁금합니다.
종결 코돈 변이에서 생길 수 있는 단백질 말단 변화에 집중해 단백질 말단부의 길이, 아미노산 구성, 소수성, 응집성 등 질병 발생과 밀접한 37가지 정밀 지표를 머신러닝에 학습시켜 TAILVAR 모델을 구축했습니다. 모델의 개발 및 성능 검증을 위해 100만 명이 넘는 대규모 공개 유전체 데이터베이스를 활용했으며, 그 결과 TAILVAR는 예측 정확도(AUROC) 0.956을 기록하며 현존하는 예측 도구 중 최고 수준의 성능을 입증했습니다.
실제 세포 실험 데이터와의 상관관계 분석에서도 가장 높은 상관성(ρ=0.379)을 나타내며, AI의 예측이 실제 생물학적 현상과 잘 맞아떨어짐을 증명했습니다. 특히 판독 불가능 상태였던 의미 불분명 변이(VUS)에 적용한 결과, 약 42%의 변이를 병원성 등으로 명확히 재분류하는 성과를 거둘 수 있었습니다.
이번 연구의 의미는 무엇인가요?
이번 연구는 종결 코돈 변이가 정말 질병을 일으키는가를 수치로 추정하고 그 결과를 실험, 임상 근거와 맞춰 확인하는 과정입니다. TAILVAR는 종결 코돈 변이에서 생길 수 있는 단백질 말단 변화에 주목해 AI 모델이 단순히 수치를 계산하는 수준을 넘어 실제 암 억제 유전자(APC, MLH1, SMAD4, BAP1 등)의 기능적 소실을 실험 데이터와 일치하게 예측해낼 수 있음을 시사합니다.
이번 연구는 그간 평가하기 어려웠던 종결 코돈 변이를 체계적으로 분석할 수 있는 새로운 기준을 제시했다는 점에 의의가 있습니다. 미지의 영역에서 발굴된 변이들이 질병에 어떻게 영향을 미치는지를 해석하려면 근거가 있어야 하고, 이를 통해 판독할 수 있습니다. 우리 몸 유전자의 전체 서열만 해도 30억 염기쌍이 되는데 거기에 A,T,G,C 네 가지 염기조합으로 가능한 경우의 수를 따지면 거의 무한대인 셈이어서 변이 효과에 대한 판독 근거를 찾기가 어렵습니다 다행히 AI 기술을 활용해 이러한 근거를 뒷받침하는 예측모델을 만들 수 있었다는 점에서 의미가 있다고 생각합니다.
이번 연구가 어떻게 활용되기를 바라시나요?
우리 연구팀이 구축한 TAILVAR 모델은 연구자들이 유전자 변이 해석과 임상 판독에 활용하도록 오픈소스 플랫폼에 공개했습니다. 이 모델은 의미 불분명 변이(VUS)를 더 빨리 재분류할 수 있다는 점에서 유용하게 활용될 수 있으므로, 새로운 질병과 유전자 사이의 관계, 그동안 몰랐던 새로운 질병을 일으키는 유전자를 찾는 데 기여할 수 있기를 바랍니다. 또한 아직 밝혀지지 않은 질병 원인 유전자와 변이 발굴을 가속화해 정밀 의료와 개인 맞춤형 치료 전략에도 도움이 될 것으로 기대합니다.

2026년 vol.126▲
 독자 퀴즈 참여
독자 퀴즈 참여 홈페이지 바로가기
홈페이지 바로가기